Investigadors de lʼInstitut de Neurociències de la UB identifiquen una proteïna implicada en els dèficits motors de la malaltia de Huntington
La malaltia de Huntington es caracteritza per una mutació en el gen de la huntingtina. Els pacients presenten moviments involuntaris, dèficits cognitius i trastorns psiquiàtrics específics de la malaltia que són fruit de la degeneració i la mort de les neurones espinals mitjanes de lʼestriat. Un treball liderat per investigadors de lʼInstitut de Neurociències de la UB (UBNeuro) ha determinat el paper tòxic de la proteïna RTP801 en aquesta patologia i, en concret, quin efecte té en els dèficits motors.
La malaltia de Huntington es caracteritza per una mutació en el gen de la huntingtina. Els pacients presenten moviments involuntaris, dèficits cognitius i trastorns psiquiàtrics específics de la malaltia que són fruit de la degeneració i la mort de les neurones espinals mitjanes de lʼestriat. Un treball liderat per investigadors de lʼInstitut de Neurociències de la UB (UBNeuro) ha determinat el paper tòxic de la proteïna RTP801 en aquesta patologia i, en concret, quin efecte té en els dèficits motors.
«Aquesta proteïna ja es va identificar en la malaltia de Parkinson, i ara hem pogut descriure-la en la malaltia de Huntington, un altre trastorn del moviment. Quan lʼexpressió dʼaquesta proteïna es regula a la baixa, prevenim lʼaparició dels dèficits motors», explica Cristina Malagelada, investigadora de lʼUBNeuro i una de les líders de la recerca, que sʼha publicat a la revista Cell Death and Disease.
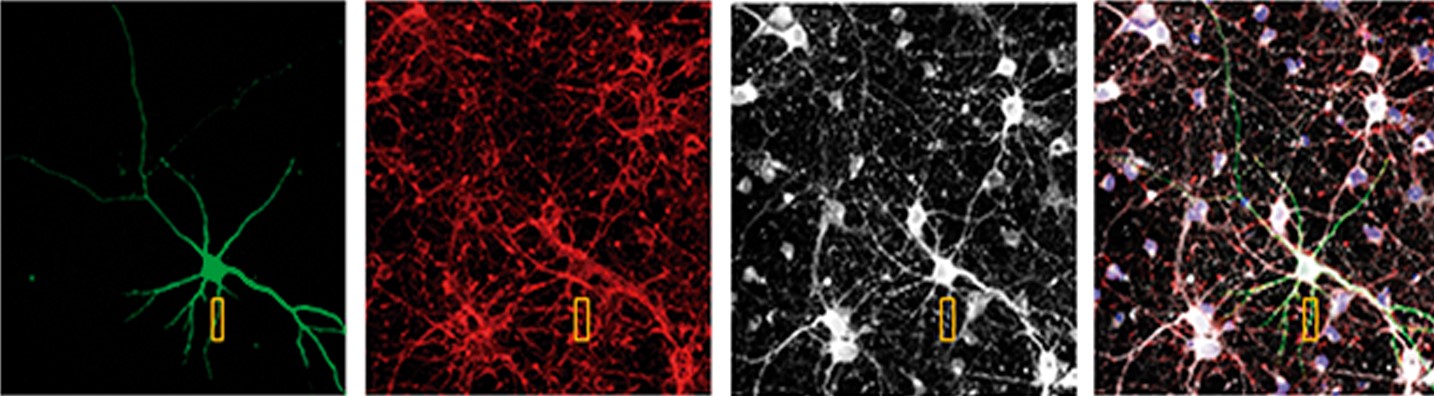
Lʼequip ha investigat si la proteïna RTP801 està involucrada en els dèficits en lʼaprenentatge motor utilitzant models de ratolí, cultius primaris de neurones i mostres de cervell post mortem de pacients de Huntington. Els resultats de lʼestudi mostren nivells alts dʼaquesta proteïna en les sinapsis de lʼestriat, tant en models de ratolí com en mostres de malalts de Huntington.
«El principal resultat del treball és que la proteïna RTP801 contribueix a lʼefecte perjudicial de la huntingtina mutada a les espines sinàptiques estriatals de ratolins que modelen la malaltia de Huntington», explica Esther Pérez-Navarro, investigadora de lʼUBNeuro i també líder de la recerca.
En lʼestudi sʼha vist que provocar una disminució de la proteïna RTP801 restaura els nivells de proteïnes crucials que estan desregulades en aquesta malaltia, com ara la cinasa Akt, RICTOR o TrkB. «Principalment, aquests resultats validen la proteïna RTP801 com a diana terapèutica en la malaltia de Huntington. El disseny de molècules o dʼestratègies terapèutiques que modulin els nivells de la proteïna RTP801 serà molt important per acabar trobant un tractament eficient per pal·liar aquesta malaltia», conclou Malagelada.
Referencia de l'article:
N. Martín-Flores et al. «Synaptic RTP801 contributes to motor-learning dysfunction in Huntington ʼs disease». Cell Death & Disease, volum 11, número d'article 569, 2020. Doi: 10.1038/s41419-020-02775-5