Un treball liderat per la UB explica la causa de la rigidesa de lʼaorta en la síndrome de Marfan
Un equip dʼinvestigadors liderat per la Universitat de Barcelona ha determinat els mecanismes bàsics que provoquen la rigidesa de lʼaorta ascendent en malalts amb la síndrome de Marfan, i en conseqüència, un aneurisma ascendent dʼaorta. Els aneurismes dʼaorta, tant els toràcics com els abdominals, són la dinovena causa de mort al món, i aquesta síndrome minoritària dʼorigen genètic és una malaltia paradigmàtica per estudiar aquest tipus de patologies.
Un equip dʼinvestigadors liderat per la Universitat de Barcelona ha determinat els mecanismes bàsics que provoquen la rigidesa de lʼaorta ascendent en malalts amb la síndrome de Marfan, i en conseqüència, un aneurisma ascendent dʼaorta. Els aneurismes dʼaorta, tant els toràcics com els abdominals, són la dinovena causa de mort al món, i aquesta síndrome minoritària dʼorigen genètic és una malaltia paradigmàtica per estudiar aquest tipus de patologies.
Les principals conclusions del treball, publicat a la revista Arteriosclerosis, Thrombosis, and Vascular Biology, expliquen la rigidesa de lʼaorta per una combinació de diversos factors. Dʼuna banda, alteracions de la matriu extracel·lular i canvis en el fenotip de les cèl·lules musculars llises; de lʼaltra, alteracions en la regulació del factor transformant de creixement beta (TGF-beta).
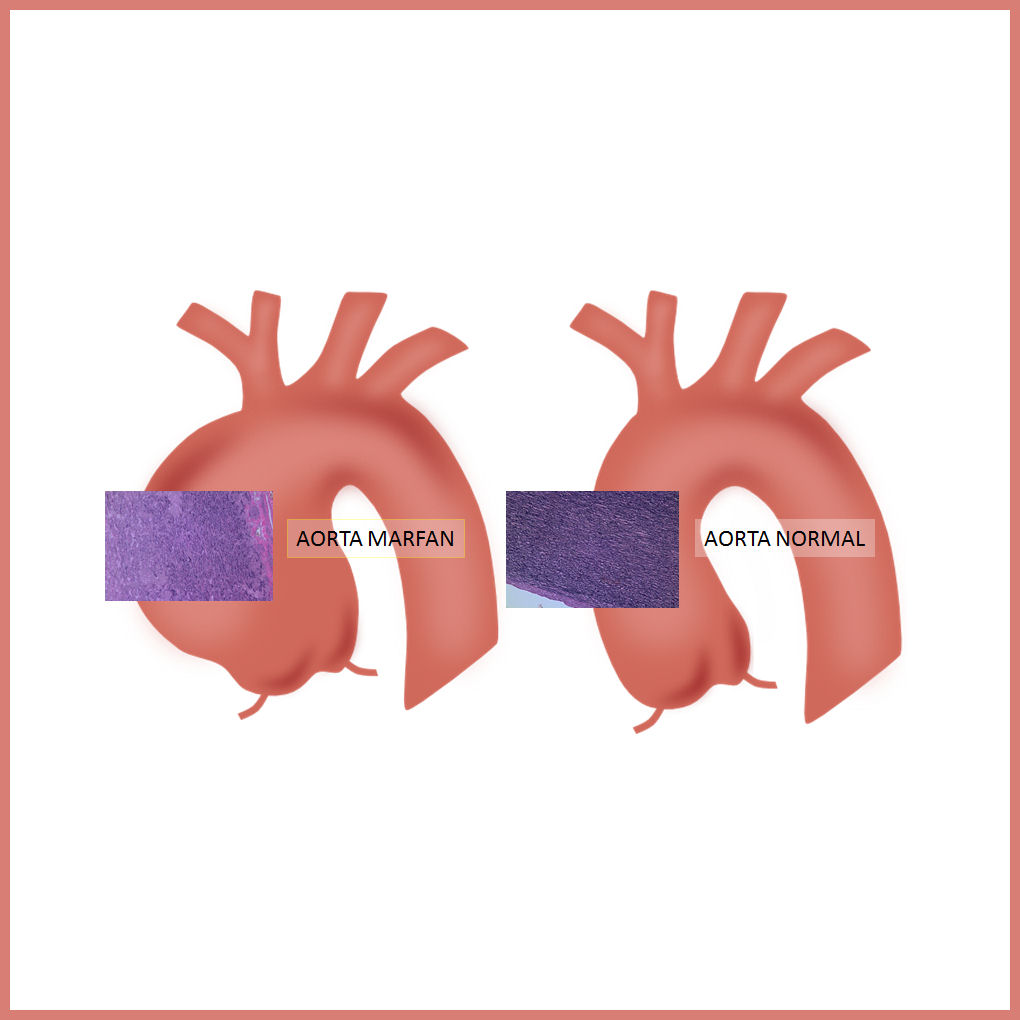
Lʼestudi mostra que el canvi en les cèl·lules i en la matriu extracel·lular explicaria la rigidesa de lʼaorta i és un marcador de lʼinici de lʼaneurisma. Tal com explica Gustavo Egea, líder de la recerca i catedràtic del Departament de Biologia Cel·lular, Immunologia i Neurociències de la UB, «aquest fenomen també es podria donar en un altre tipus dʼaneurismes dʼaorta que no tenen una causa genètica. Per tant, aquest podria ser un mecanisme general que en el cas de Marfan té lloc de manera més accelerada». El doctor Egea també és membre de lʼInstitut de Nanociència i Nanotecnologia (IN2UB) i de lʼInstitut dʼInvestigacions Biomèdiques August Pi i Sunyer (IDIBAPS).
La recerca sʼha dut a terme en estreta col·laboració amb Isabel Fabregat, professora del Departament de Ciències Fisiològiques II de la UB i investigadora de lʼInstitut dʼInvestigacions Biomèdiques de Bellvitge (IDIBELL), i ha utilitzat mostres de pacients de Marfan intervinguts per fer-los una reparació dʼaorta. Se nʼhan pres mostres tant de la zona afectada com de zones més allunyades. Aquestes mostres sʼhan pogut comparar amb dʼaltres de donants de cor, i a partir de lʼanàlisi cel·lular, molecular i biofísic sʼha pogut estudiar, tant en el teixit com en el cultiu de cèl·lules, els mecanismes bàsics que es veuen alterats en la malaltia.
En els malalts de Marfan té lloc una mutació genètica en el gen que codifica per a la proteïna fibrilina I, que causa la malformació i lʼassemblatge de les fibres elàstiques. Aquestes, conjuntament amb les cèl·lules musculars llises, constitueixen la part més important de la paret de lʼaorta toràcica. El treball mostra que les cèl·lules musculars llises que hi ha al mig dʼaquestes fibres adquireixen un fenotip molt més contràctil del que és normal, i això les fa més rígides.
La síndrome de Marfan és una malaltia genètica causada per la mutació dʼun gen que codifica per a la proteïna fibrilina I, un dels dos components principals de les fibres elàstiques que formen el teixit conjuntiu. Com a conseqüència dʼaquesta mutació, lʼassemblatge de les fibres elàstiques als teixits es fa malament, i per tant, la seva funció de distensió i relaxació es perd i els teixits es fan malbé de manera accelerada.
Tots els teixits on hi ha moltes fibres elàstiques seʼn veuen afectats, com ara la pell, on surten estries, o el cristal·lí de lʼull, que es desplaça i causa ceguesa. De totes aquestes disfuncions, la més important és lʼafebliment accelerat de lʼaorta ascendent, que dóna lloc a lʼaneurisma aòrtic i la posterior dissecció de lʼaorta.
El conjunt dʼaquestes manifestacions clíniques és el que es coneix com a síndrome de Marfan, que tot i ser una malaltia minoritària té una prevalença elevada dʼ1/5.000 pacients i és difícil de diagnosticar.
Els malalts de Marfan són persones molt altes amb les extremitats desproporcionadament llargues. La vida mitjana dels malalts és dʼuns 40 anys, i prop del 50 % de la població que té la malaltia no està diagnosticada. A Catalunya hi pot haver a lʼentorn de 5.000 persones que la pateixen.
El diagnòstic es fa mitjançant un estudi de les manifestacions clíniques que tenen una puntuació. En cas de dubte es pot fer una anàlisi genètica.
Aquest treball és altament transversal. Hi han participat també, la Unitat de Marfan de Madrid i un grup de cirurgians de lʼHospital Clínic de Barcelona, de l'Hospital Doce de Octubre de Madrid i de lʼHospital de Bellvitge, en què també hi han intervingut patòlegs. Ha estat igualment rellevant la participació dʼinvestigadors de lʼInstitut de Bioenginyeria de Catalunya liderats per Daniel Navajas. En lʼarticle també hi ha col·laborat Hal C. Dietz, que el 1991 va descobrir que la mutació en la fibrilina I causa la malaltia.
E. Crosas-Molist, T. Meirelles, J. López-Luque, C. Serra-Peinado, J. Selva, L. Caja, D. Gorbenko del Blanco, J. J. Uriarte, E. Bertran, Y. Mendizábal, V. Hernández, C. García-Calero, O. Busnadiego, E. Condom, D. Toral, M. Castellà, A. Forteza, D. Navajas, E. Sarri, F. Rodríguez-Pascual, H. D. Dietz, I. Fabregat, G. Egea. «Vascular smoooth muscle cell phenotypic changes in patients with Marfan syndrome» Arteriosclerosis, Thrombosis, and Vascular Biology. 2015. DOI: 10.1161/ATVBAHA.114.304412