Advancing research into Schaaf-Yang syndrome, a rare disease that causes intellectual disability and congenital malformations
Mutations in the MAGEL2 gene, which cause Schaaf-Yan syndrome (SYS) —an ultra-rare disease that affects neuronal and cognitive development— generate truncated, non-functional proteins that tend to accumulate in the cell nucleus. Moreover, this progressive accumulation of abnormal proteins could cause a toxic effect in patients affected by the syndrome, who suffer congenital malformations, intellectual disability, alterations in facial features, sleep apnoea and joint contractures.
Mutations in the MAGEL2 gene, which cause Schaaf-Yan syndrome (SYS) —an ultra-rare disease that affects neuronal and cognitive development— generate truncated, non-functional proteins that tend to accumulate in the cell nucleus. Moreover, this progressive accumulation of abnormal proteins could cause a toxic effect in patients affected by the syndrome, who suffer congenital malformations, intellectual disability, alterations in facial features, sleep apnoea and joint contractures.
These advances in SYS research appear in a study published in the Journal of Medical Genetics. The study was led by a team from the Faculty of Biology and the Institute of Biomedicine of the University of Barcelona (IBUB), the Sant Joan de Déu Research Institute (IRSJD) and the Rare Diseases Networking Biomedical Research Centre (CIBERER). This team is also the author of the publication of the first clinical guide on Schaaf-Yang syndrome (Journal of Medical Genetics, 2022), aimed at healthcare professionals and families of children affected by this pathology.
A better understanding of the function, genetic variants and impact of nuclear retention of the MAGEL2 protein will open new ways to design patient-specific gene therapies to prevent the synthesis of the altered protein and address SYS, a disease without treatment.
Genetic mutations leading to truncated proteins
The MAGEL2 gene is located on chromosome 15, is expressed in the nervous system and produces the MAGEL2 protein, which is involved in the retrograde transport and recycling of proteins in the cell cytoplasm of neurons. To date, more than eighty mutations in the MAGEL2 gene have been documented in the scientific literature, some of which are found repeated among patients. Currently, it is estimated that there are about 250 people diagnosed with Schaaf-Yang syndrome worldwide.
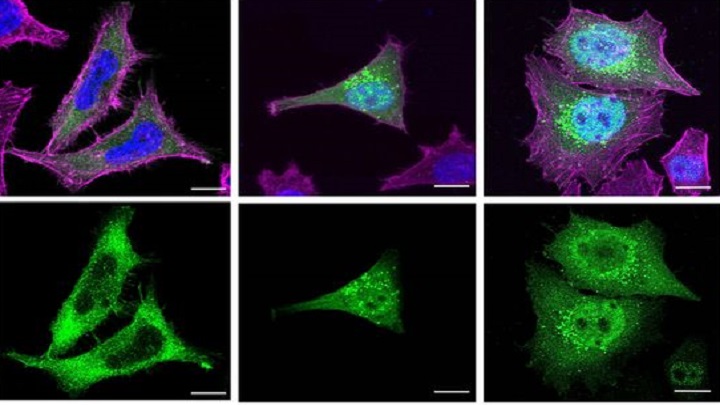
The new study, conducted with human cells in vitro, shows how almost all truncated proteins associated with Schaaf-Yang syndrome lose part of their molecular structure due to genetic mutations. “Functional MAGEL2 proteins have a complete molecular structure that allows them to interact with other proteins and carry out their normal biological functions. They are usually found in specific locations within the cytoplasm of the cell, mainly in subcellular compartments related to the transport and recycling of proteins”, explains Susanna Balcells, professor at the UB’s Department of Genetics, Microbiology and Statistics, and coordinator of the study. “In contrast — she continues — truncated proteins are shorter versions of the MAGEL2 protein, as they have been affected by genetic mutations. Therefore, truncated proteins lack certain regions necessary to function correctly in the cell”.
Due to genetic mutations, truncated proteins lose key structural domains, such as the MAGE homology domain, which is crucial for interactions with other proteins. “The absence of this domain could prevent these essential interactions for the correct functioning of MAGEL2, such as its role in retrograde transport and protein recycling”, explains Roser Urreizti.
When toxic proteins accumulate in the cell nucleus
Truncated proteins tend to accumulate inside the cell nucleus, and this could further aggravate the symptomatology of people affected by Schaaf-Yang. Mónica Centeno, “it is likely that, in a real cellular context, some of the truncated proteins synthesised can also be found in specific locations within the cytoplasm, such as endosomes, for example. However, as their structure is altered, they might not be able to perform their normal functions correctly”.
The severity of clinical manifestations in those affected by Schaaf-Yang syndrome may be related to the accumulation of altered proteins in the cell nucleus. “In other words, the mutations that cause more severe symptoms also cause the truncated MAGEL2 protein to accumulate more in the nucleus. This could be explained by the fact that a higher accumulation of truncated proteins in the nucleus could be interfering with important nuclear processes and affect the functioning of the cell under normal conditions to a greater degree,” says Raquel Rabionet.
Searching for treatments to address Schaaf-Yang syndrome
Cellular mechanisms that drive protein degradation — for example, the ubiquitin-proteasome system — could help reduce the negative effects of truncated proteins and contribute to mitigating the progression of the pathology. However, if the rate of protein production exceeds the degradation capacity of the cell, aberrant proteins may escape degradation and continue to exert toxic effects. The expert Aina Prat says that “in this regard, we have found that normal and truncated MAGEL2 proteins have very similar half-lives. Therefore, truncated MAGEL2 would be stable in the cell, where it could be exerting toxic effects”.
“If we knew how truncated MAGEL2 proteins alter cell function, we could develop strategies to promote the degradation of these toxic proteins, restore cell function or compensate for the metabolic and signalling dysfunctions caused by their accumulation”, concludes the team, which will drive further research to contribute to the development of innovative treatments for people affected by SYS.
Reference article:
Centeno, Mónica; Alcaide-Consuegra, Estefanía; Gibson, Sophie; Prat-Planas, Aina; Gutiérrez-Ávila, Juan D.; Grinberg, Daniel; Urreizti, Roser; Rabionet, Raquel; Balcells, Susanna. “Subcellular localisation of truncated MAGEL2 proteins: insight into the molecular pathology of Schaaf-Yang syndrome”. Journal of Medical Genetics, March 2024. DOI: 10.1136/jmg-2024-109898
Multimedia gallery

From left to right, Mónica Centeno, Aina Prat, Juan Diego Gutiérrez, Susanna Balcells and Raquel Rabionet, members of the UB, the BUB, the IRSJD and the CIBERER.
